|
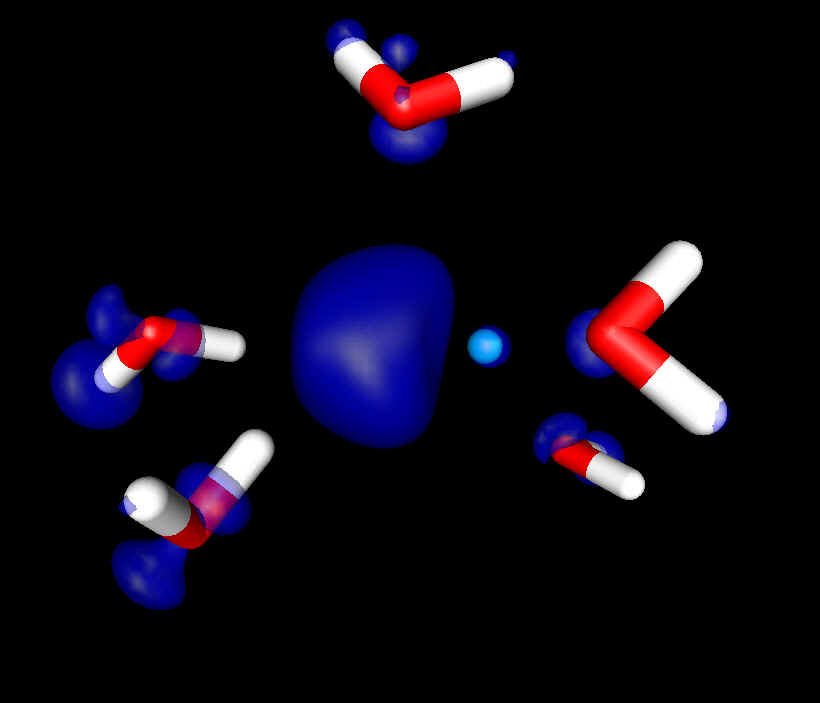
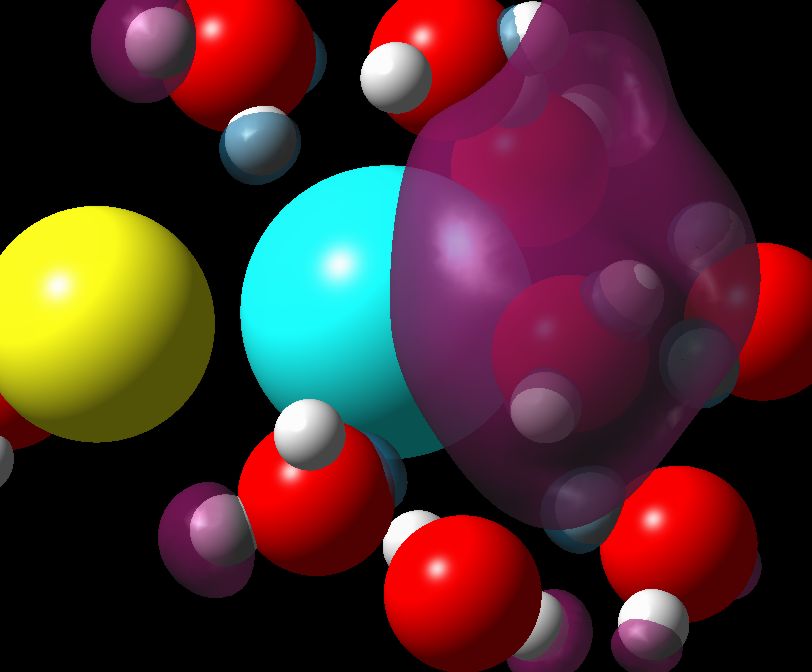
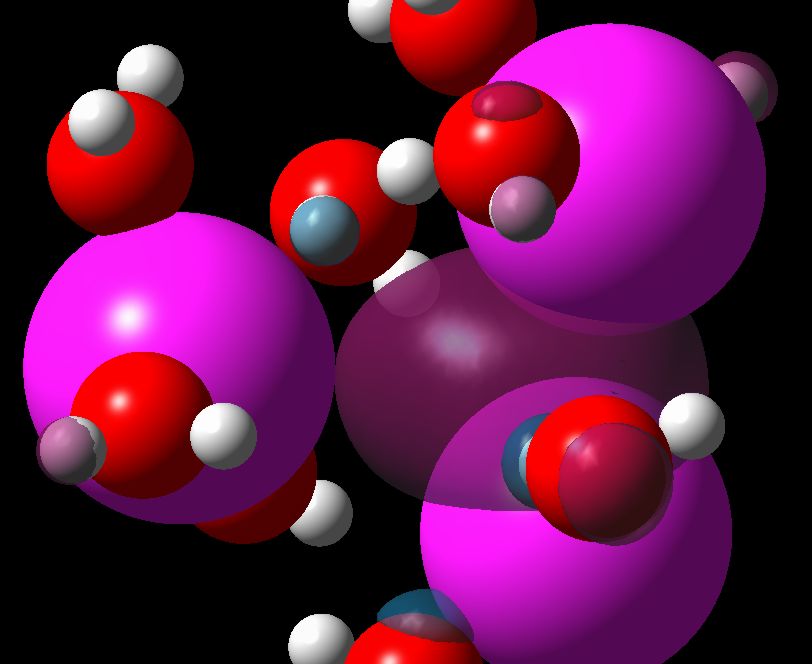
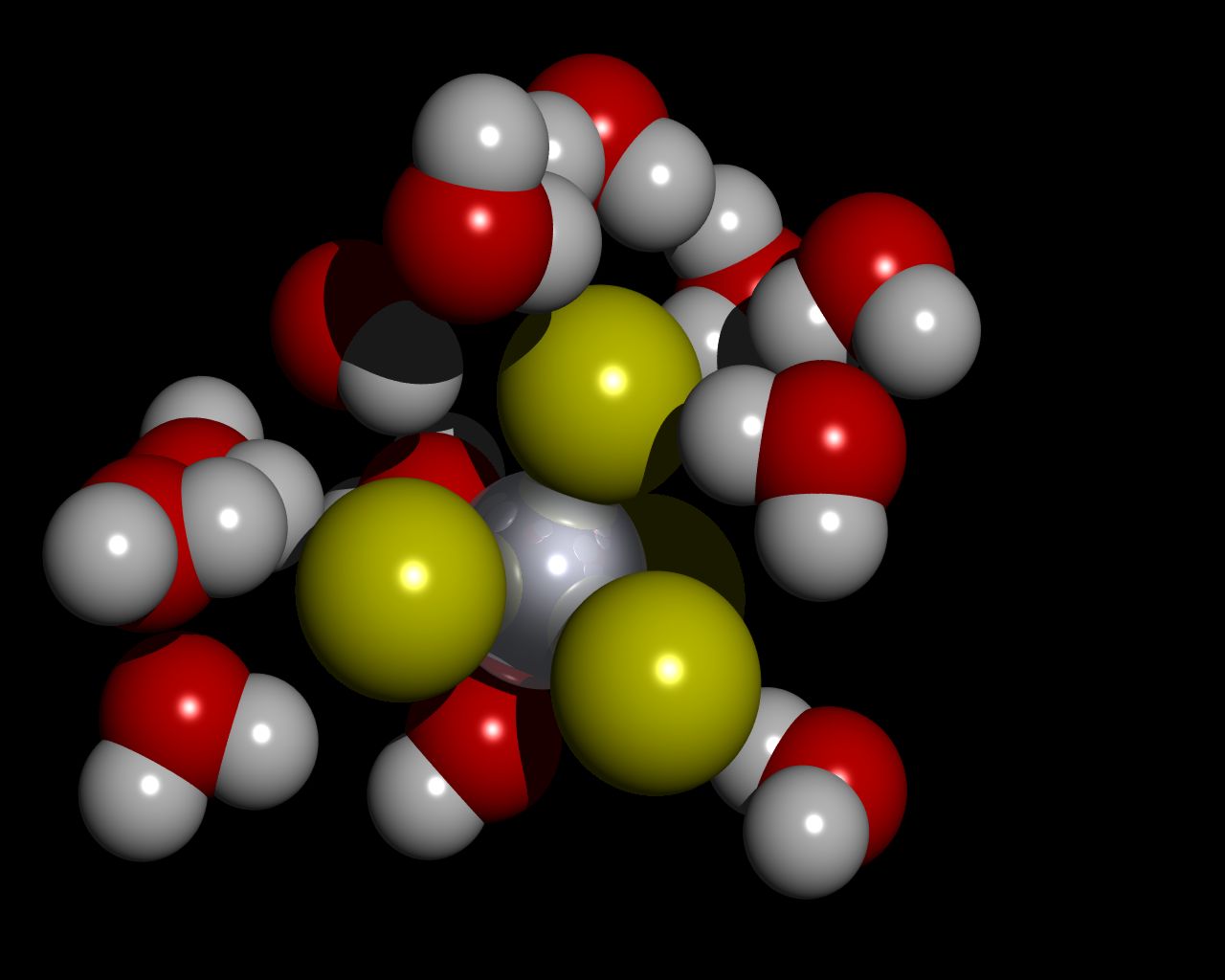
Excess electron localization in concentrated ionic solutions. A computer simulation research The calculations were split into three stages: a) Classical Molecular Dynamics simulation of the aqueous solutions structure, b) Quantum Molecular Dynamics of the excess electron localization in the solutions and c) Quantum Mechanical simulations of the excess electron absorption spectrum. Molecular Dynamics simulations of LiCl, NaCl, MgCl2 and NaOH aqueous solutions were performed with the flexible model of water molecule. Thanks to the analysis of the structural and dynamical properties of the solutions, the regions of the crystalline-order at high concentrations have been found. The subsets originating from equilibrated solutions were selected to perform Quantum Car-Parinello Molecular Dynamics of the excess electron localization. The calculations were performed on the DFT level using plane wave basis set, Goedecker-type pseudopotentials and LSD approximation. Obtained electron spin densities were analyzed using pair correlation functions. Clusters composed of the localized electron and its surrounding were selected to calculate the absorption spectrum with time-dependent, non-local DFT method. Three electron-trapping sites have been found in aqueous ionic solutions: purely water environment (responsible for evis absorption band), cationic traps (responsible for eir band) and occasionally, traps of the “F‑centre” type. Committee for Scientific Research (KBN) grant nr: KBN 3T09A 06617 |
|||
|
|
|
|
|